Migration and population structure#
The genomic transmission graph can be broken down into many local subpopulations that together make up a metapopulation. Here we examine a local subpopulation that is embedded within a much larger metapopulation.
\(m\) is the probability that a host within the local subpopulation acquired their infection from the metapopulation, and the number of such hosts per generation is \(N_m\) = \(mN_h\). These could be immigrants from the metapopulation or local residents who have been travelling outside the local area.
We must modify our Markov chain simulation of coalescence times to account for migration. Within coalestr this is split this into two components, with one transmission probability matrix for the subpopulation and another transition probability matrix for the metapopulation.
In coalestr we can examine the effects of migration within a hierarchical population structure as follows:
We create a metapopulation by specifying its transmission history
pass this to
coalestr.Population()and give the metapopulation a namerun
get_coalescent()for the metapopulationrun
get_diversityfor the metapopulation
We then create a subpopulation by specifying its transmission history and the number of migrant hosts per generation, \(N_m\).
pass to
coalestr.Population()the transmission history and the name of the metapopulationrun
get_coalescent()for the subpopulationrun
get_diversityfor the subpopulation
\(F_{ST}\) is automatically calculated and returned as an attribute of the subpopulation.
!pip install coalestr
from coalestr import cs
import matplotlib.pyplot as plt
import coalestr as cs
# define the metapopulation
metapop_history = [[100000, 3000, 10, 1, 0]]
metapop = cs.Population(metapop_history)
metapop.get_coalescent()
metapop.get_diversity()
Observation time. Events captured. Mean coalescence time
beho wiho beho wiho
0 99.7 99.7 16867.3 15476.3
Observation time. Nucleotide diversity Haplotype homozygosity
beho wiho beho wiho
0 3.71e-04 3.40e-04 4.41e-03 8.47e-02
# define the subpopulation
Nm_list = [0, 0.1, 1, 10, 30]
duration = 1000
Nh = 30
Q = 10
X = 0.5
for Nm in Nm_list:
subpop_history = [[duration, Nh, Q, X, Nm]]
subpop = cs.Population(subpop_history, metapopulation = metapop)
subpop.get_coalescent(show = False)
subpop.get_diversity(show = False)
beho_nuc_div = subpop.diversity[0,1]
beho_hap_hom = subpop.diversity[0,3]
fst = subpop.diversity[0,6]
print("Nm = {0:4.2f}: nuc div = {1:.2e}, hap hom = {2:4.2f}, Fst = {3:4.2f}".format(
Nm,
beho_nuc_div,
beho_hap_hom,
fst))
Nm = 0.00: nuc div = 2.42e-06, hap hom = 0.41, Fst = 0.99
Nm = 0.10: nuc div = 1.54e-04, hap hom = 0.32, Fst = 0.59
Nm = 1.00: nuc div = 3.26e-04, hap hom = 0.10, Fst = 0.12
Nm = 10.00: nuc div = 3.67e-04, hap hom = 0.01, Fst = 0.01
Nm = 30.00: nuc div = 3.70e-04, hap hom = 0.01, Fst = 0.00
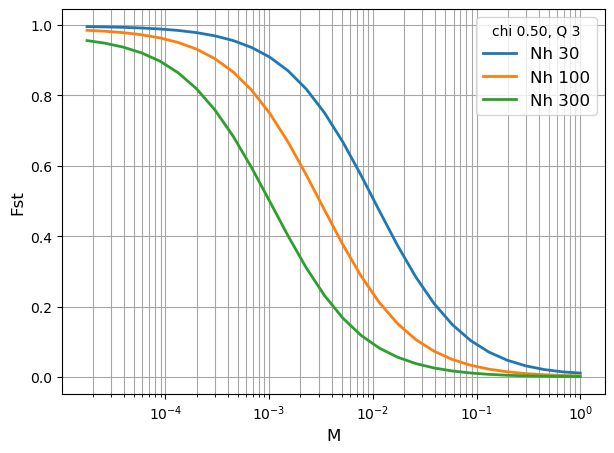
# define a set of subpopulations with different values of Nh
N_list = [30, 100, 300]
Q = 3
chi = 0.5
duration = 10000
iterations = 28
fig, ax = plt.subplots(figsize=(7,5))
for i in range(len(N_list)):
Nh = N_list[i]
M_list = []
Fst = []
for j in range(iterations):
M = 1.5 ** (-j)
Nm = Nh * M
subpop_history = [[duration, Nh, Q, chi, Nm]]
subpop = cs.Population(subpop_history, metapopulation = metapop)
subpop.get_coalescent(show = False)
subpop.get_diversity(show = False)
M_list.append(M)
Fst.append(subpop.diversity[0, 6])
ax.plot(M_list, Fst, linewidth = 2, label = 'Nh {0:.0f}'.format(Nh))
ax.legend(title = 'chi {0:.2f}, Q {1:.0f}'.format(chi, Q), frameon=True, fontsize=12)
ax.set_xlabel("M", fontsize=12)
ax.set_ylabel("Fst", fontsize=12)
ax.set_xscale('log',base=10)
ax.grid(visible=True, which='both', color='0.65', linestyle='-')
plt.show()